Así como la masa, la energía puede considerarse a ser conservada en todos excepto los procesos nucleares.
La conservación de la energía, sin embargo, difiere de la masa en que la energía puede ser generada (o consumida) en un proceso químico. La materia puede cambiar de forma, pueden formarse nuevas especies químicas por reacción, pero el flujo total de masa que entra a una unidad de proceso debe ser igual al flujo de salida en el estado estacionario. Lo mismo no sucede con la energía. La entalpía total de las corrientes de salida no será igual a la de las corrientes de entrada si se genera o consume energía en el proceso; como el que se debe al calor de reacción. La energía puede existir en varias formas: calor, energía mecánica, energía eléctrica, y ellas formaran el total de energía.
En el diseño de procesos, los balances de energía se hacen para determinar los requerimientos de energía del proceso: el calentamiento, enfriamiento y potencia requerida. En una planta en operación, un balance de energía sobre la planta mostrará la cantidad de consumo de energía y sugerirá áreas para conservación y ahorro.
En este capítulo se analizan brevemente los fundamentos de los balances de energía, y se dan ejemplos para ilustrar el uso de los balances de energía en el diseño de procesos.
5.1. CONSERVACIÓN DE LA ENERGIA
Así como para la materia, se puede escribir una ecuación general para la conservación de la energía.
Energía que sale = Energía que entra + Generación – Consumo + Acumulación.
Este es un enunciado de la primera ley de la termodinámica.
– Un balance de energía puede escribirse para cualquier etapa del proceso.
– La reacción química liberará energía (exotérmica) o consumirá energía (endotérmica).
– Para procesos al estado estacionario la acumulación de masa y energía será cero.
– La energía puede existir en muchas formas e esto, para algunos, casos harán los balances de energía mas complejos que los balances de materia.
5.2. FORMAS DE ENERGÍA (POR UNIDAD DE MASA DEL MATERIAL)
5.2.1 Energía potencial
Energía debida a la posición
Energía potencial = gz (5.1)
donde: z = altura sobre un punto de referencia, m
g = aceleración gravitacional, (9,81 m/s2)
5.2.2 Energía cinética
Energía debida al movimiento
![]()
donde: u = velocidad, m/s
5.2.3 Energía interna
Energía asociada con el movimiento molecular. La temperatura T de un material es una medida de su energía interna U
U = f(T) (5.3)
5.2.4 Trabajo
Se realiza trabajo cuando una fuerza actúa a lo largo de una distancia
![]()
donde: F = fuerza, N
x y L = distancia, m
El trabajo realizado sobre un sistema por los alrededores es convencionalmente tomado como negativo; el trabajo realizado por el sistema sobre los alrededores como positivo.
Cuando el trabajo se debe a un cambio en la presión o volumen
![]()
donde: P = presión, Pa (N/m2)
v = volumen por unidad de masa, m3/kg
Para integrar esta función deben conocerse las relaciones entre la presión y el volumen. En el diseño de procesos, frecuentemente es necesario estimar el trabajo realizado durante la compresión o expansión de un gas. Se puede hacer un estimado preliminar asumiendo ya sea una expansión adiabática reversible (isentrópica) o una expansión isotérmica, dependiendo de la naturaleza del proceso.
Para expansión isotérmica (expansión isotérmica (expansión a temperatura constante)
Pv = constante
Para expansión adiabática reversible (sin intercambio de calor con los alrededores)
pvg = constante
donde: g = razón de capacidades caloríficas, Cv/Cv.
5.2.5 Calor
La energía es transferida ya sea como calor o trabajo. Un sistema no contiene “calor”, pero la transferencia de calor o trabajo a un sistema cambia su energía interna. El calor tomado por un sistema desde sus alrededores es convencionalmente tomado como positivo y el que cede el sistema como negativo.
5.2.6 Energía eléctrica
Las formas de energía eléctrica y mecánica son incluidas en el término del trabajo en un balance de energía. La energía eléctrica será significante solamente en los balances de energía para procesos electroquímicos.
5.3. EL BALANCE DE ENERGÍA
Considerando un proceso al estado estacionario representado por la fig. 5.1. La ecuación de conservación se puede escribir para incluir las diferentes formas de energía.

Fig. 5.1 Proceso general al estado estacionario
Por unidad de masa de material:
![]()
Los sufijos 1 y 2 representan los puntos de entrada y salida respectivamente. Q es el calor transferido a través de los límites del sistema; positivo para el calor entrando al sistema, negativo para el calor saliendo del sistema. W es el trabajo realizado por el sistema, positivo para el trabajo yendo desde el sistema hacia los alrededores, y negativo para el trabajo entrando al sistema desde los alrededores.
La ecuación 5.6 es una ecuación general para sistemas con flujo al estado estacionario.
En procesos químicos, los términos de energía cinética y energía potencial son usualmente pequeños comparados con los términos de calor y trabajo, y pueden normalmente ser despreciados.
Es conveniente, y provechosos, tomar los términos U y pV juntos; definiendo el término entalpía, el símbolo usual es H, así:
H = U + PV
La entalpía es una función de la temperatura y presión. Valores para las sustancias mas comunes han sido determinadas experimentalmente y son dadas en los diferentes “hanbooks”. Así mismo, la entalpía puede calcularse a partir de datos de calor específico y calor latente.
Si los términos de energía cinética y energía potencial se desprecian, la ecuación 5.6 se reduce a:
H2 – H1 = Q – W (5.7)
Esta ecuación simplificada es usualmente suficiente para estimar los requerimientos de calentamiento y enfriamiento de las diferentes unidades de operación incluidas en los procesos químicos.
Como los términos dependientes del flujo han sido omitidos, esta ecuación simplificada es aplicable tanto para sistemas estáticos (sin flujo) y sistemas con flujo. Y puede usarse para estimar los requerimientos de energía para los procesos “batch”.
Para muchos procesos el término del trabajo será cero, o muy pequeño y la ecuación 5.7 se reduce a una ecuación de simple balance de calor.
Q = H2 – H1 (5.8)
Cuando se genera calor en el sistema; por ejemplo en un reactor químico.
Q = Qp + Qs (5.9)
donde: Qs = calor generado en el sistema, si se libera calor (proceso exotérmico), Qs es
tomado como positivo, y si se absorbe calor (proceso endotérmico) es
tomado como negativo.
Qp = calor adicionado al proceso por el sistema para mantener la temperatura
requerida del sistema.
De aquí:
Qp = H2 – H1 – Q1
H1 = entalpía de la corriente de entrada
H2 = entalpía de la corriente de salida.
Ejemplo 5.1
Balance sin reacción química. Estimar las cantidades de vapor y agua requeridas para la columna de destilación mostrada en la figura.
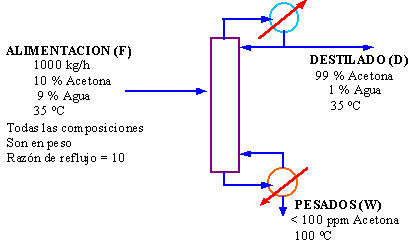
En la planta se dispone de vapor seco saturado a 25 psig (276 kN/m2), el agua de enfriamiento puede aumentar su temperatura en 30 oC. La columna opera a 1 bar
Solución
Balance de materiales
Es necesario hacer un balance de materiales para determinar los flujos de productos en el tope y el fondo.
Balance de acetona, despreciando las pérdidas de acetona en el fondo.
100 x 0,1 = D x 0,99
Destilado, D = 101 kg/h
Pesados, W = 1000 – 101 = 899 kg/h
Balance de energía
Las energías cinética y potencial de las corrientes del proceso son pequeñas y pueden despreciarse.
Tomando los límites del primer sistema que incluyan al hervidor y al condensador.
Entradas: calor que entra al hervidor QB + calor sensible de la alimentación HF
Salidas: enfriamiento para el condensador QC + calor sensible de los productos del tope y el fondo HD + HW.

Las pérdidas de calor desde el sistema serán pequeñas si la columna y los intercambiadores están aislados convenientemente (típicamente menor a 5 por ciento) y será despreciado.
Bases: 25 oC, 1 hr.
Capacidades caloríficas promedio:
Acetona 25 oC a 35 oC = 2,2 kJ/kg oK
Agua 25 oC a 100 oC = 4,2 kJ/kg oK
Capacidades caloríficas promedio de las corrientes:
Alimentación, 10 por ciento de acetona = 0,1 x 2,2 + 0,9 x 4,2 = 4,00 kJ/kg oK
Tope, 99 por ciento de acetona, tomado como acetona, 2,2 kJ/kg. oK
Pesados, como agua, 4,2 kJ/kg oK
QC debe determinarse mediante un balance alrededor del condensador y hervidor.

Razón de reflujo
R = L/D = 10
L = 10 x 101 = 1010 kg./h
V= L + D = 1111 kg./h
De los datos para la ebullición de una mezcla con 99 por ciento de acetona 1 por ciento de agua = 56.5 oC
Al estado estacionario: entradas = salidas
Hv = HD + HC + QC
QC = Hv – HD – HL
Asumiendo condensación completa
Entalpía del vapor HV = calor latente + calor sensible
Aquí hay dos caminos para calcularla entalpía específica del vapor a su punto de ebullición.
(1) Calor latente de vaporización a la temperatura base + calor sensible para calentar el vapor hasta el punto de ebullición.
(2) Calor latente de vaporización al punto de ebullición + calor sensible para llevar el líquido al punto de ebullición.
Teniendo valores del calor latente de acetona y agua como funciones de la temperatura, se usará el segundo método.
Calor latente de acetona a 56,5 oC (330 oK) = 620 kJ/kg.
Calor latente del agua a 56,5 oC (330 oK) = 2500 kJ/kg.
Luego:
HV = 1111 [(0.001 x 2500 + 0.99 x 620) + (56,5 – 25) 2,2] = 786 699 kJ/h
Las entalpías de los productos del tope y del reflujo serán cero, ya que ellos están a la temperatura base. Ambos son líquidos, y el reflujo estará a la misma temperatura del producto.
De aquí
QC = HV = 786 699 kJ/kg. (218,5 kW)
QB es determinado por un balance sobre el sistema total
Entrada = Salida
QB + HF = QC + HD + HW
HF = 1000 x 4,00 (35 – 25) = 40 000 kJ/h
HW = 899 x 4,2 (100 – 25) = 283 185 kJ/h
(El punto de ebullición de los productos es tomado como 100 oC)
de aquí:
QB = QC + HD + HW – HF
= 786 699 + 283 185 + 0 – 40 000
= 1029 884 kJ/h (286,1 kW)
QB es suministrado por el vapor condensando
Calor latente del vapor = 2730 kJ/kg. a 275,8 kN/m2
![]()
QC es removido por el agua de enfriamiento con un incremento en su temperatura de 30 oC
QB = flujo de agua x 30 x 4,2
![]()
5.4. CALCULO DE LA ENTALPÍA ESPECIFICA
Valores tabulados de entalpía están disponibles solamente para los materiales mas comunes. En ausencia de datos publicados, se pueden usar las siguientes expresiones para estimar la entalpía específica (entalpía por unidad de masa)
Para materiales puros, sin cambio de fase
donde: HT = entalpía específica a la temperatura T,
Cp = capacidad calorífica específica del material, a presión constante
Td = temperatura de referencia.
Si toma lugar un cambio de fase entre la temperatura de referencia y la especificada, el calor latente del cambio de fase es adicionado al cambio de calor sensible calculado por la Ecuación. 5.11.
El cálculo del calor sensible es entonces realizado en dos partes.
donde: TP = temperatura del cambio de fase
Cp1 = capacidad calorífica específica de la primera fase antes de Tp
Cp2 = capacidad calorífica específica de la segunda fase después de Tp
El calor específico a presión constante varía con la temperatura y para usar las Ecuaciones 5.11 y 5.12 se deben aprovechar valores de Cp como función de la temperatura. Para sólidos y gases, el Cp es usualmente expresado como una ecuación empírica de potencias en serie.
Cp = a + bT + cT2 + dT3 (5.13 a)
ó Cp = a + bT + cT-1/2 (5.13 b)
Las escalas absoluta (oK) o relativa (oC) de temperatura se pueden usar cuando las relaciones son de la forma dada por la ecuación 5.13 a. Para la ecuación 5.13 b debe usarse la temperatura absoluta.
Ejemplo 5.2
Estimar la entalpía específica de alcohol etílico a 1 bar y 200 oC, tomando como temperatura de referencia 0 oC
Cp liquido a 0 oC = 24,65 cal/mol oC
100 oC = 37,96 cal/mol oC
Cp gas (t en oC) = 14,66 + 3,758 x 10-2 t – 2,09 x 10-5 t2 + 4,74 x 10-9 t3 cal/mol
Punto de ebullición del alcohol etílico a 1 bar = 78,4 oC
Calor latente de vaporización = 9,22 k cal/mol
Solución
Nota: como los datos tomados de la literatura están en cal/mol, los cálculos son hechos en estas unidades y los resultados convertidos al SI.
Como se dan datos de la exacta variación del Cp del liquido con la temperatura, usar una ecuación de la forma Cp = a + b t, calculando a y b de los datos dados; esto debe ser exacto para el rango de temperatura necesario.
a = valor de Cp a 0 oC
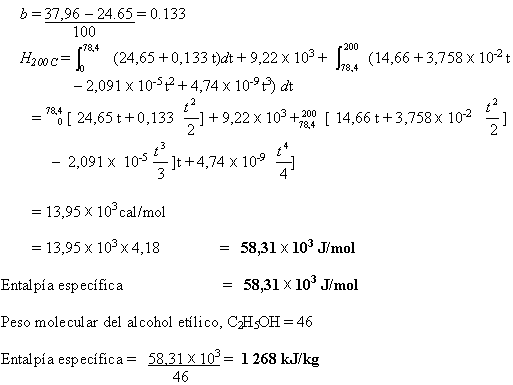
5.5. CAPACIDADES CALORÍFICAS PROMEDIO
El uso de las capacidades caloríficas promedio frecuentemente facilita el calculo de los cambios de calor sensible; la capacidad calorífica promedio en el rango de temperaturas t1 a t2 se define por la siguiente ecuación:

Valores de calores específicos promedio están tabulados en varios “handboocks”. Si los calores están calculados a partir de una temperatura de referencia, tr entonces el cambio de entalpía entre las temperaturas t1 y t2 está dada por
DH = Cpm t2 (t2 – tr) – Cpm t1 (t1 – tr) (5.15)
donde : tr es la temperatura de referencia a la cual se han calculado los valores de Cpm.
Si Cp está expresado como un polinomio de la forma Cp = a + b t + c t2 + d t3, entonces la forma integrada de la ecuación 5.14 será:

donde: t es la temperatura a la cual se requiere Cpm.
Si la temperatura de referencia es tomada como 0 oC, la ecuación 5.16 se reduce a:
![]()
y el cambio de entalpía de t1 a t2 está dado por:
DH = Cpm, t2 t2 – Cpm, t1 t1 (5.18)
Ejemplo 5.3
El gas que sale de una cámara de combustión tiene la siguiente composición: CO2 7,8; CO 0,6; O2 3,4; H2O 15,6; N2 72,6; todos son porcentajes en volumen. Calcular el calor removido si los gases son enfriados desde 800 a 200 oC.
Solución
Capacidades caloríficas medias (J/mol oC), tomando como temperatura de referencia 0 oC.
oC N2 O2 CO2 CO H2O
200 29,24 29,95 40,15 29,52 34,12
800 30,77 32,52 47,94 31,10 37,38
Calor extraído de los gases al enfriarlos desde 800 a 200 oC
= Mc (Cpm 800 x 800 – Cpm 200 x 200)
donde: Mc = moles de cada componente.
Base: 100 moles de gas (como el análisis es en volumen), sustituyendo se tiene:

5.6. EFECTO DE LA PRESIÓN SOBRE LA CAPACIDAD CALORÍFICA
Los datos de las capacidades caloríficas dados en los “handbooks”, son usualmente para el estado del gas ideal. La ecuación 5.13 a debería escribirse como:
Cpo = a + b T + c T2 + d T3 (5.19)
donde el supercero se refiere al estado de gas ideal.
Los valores para el comportamiento ideal, pueden usarse para el comportamiento real a bajas presiones. A presiones altas, el efecto de la presión sobre el calor específico puede ser muy apreciable.
Edmister (1948) publicó una correlación generalizada mostrando la corrección debido a la presión a condiciones isotérmicas para gases reales como función de la presión reducida y la temperatura y se conoce como la carta de Edmister’s (Sterbacek et al). 1979), la cual está basada en hidrocarburos, pero que puede usarse para otros materiales para dar una indicación del probable error si se usan valores de calores específicos de gases ideales sin corregirlos.
El método es ilustrado en el Ejemplo 5.4
Ejemplo 5.4
La capacidad calorífica del etileno para el estado ideal está dada por la ecuación:
Cp = 3,95 + 15,6 x 10-2 T – 8,3 x 10-5 T2 + 17,6 x 10-9 T3 J/mol K
Estimar el valor a 10 bar y 300 K.
Solución
Etileno: presión crítica 50,5 bar
Temperatura crítica 283 K
Cp = 3,95 + 15,6 x 10-2 x 300 – 8,3 x 10-5 x 3002 + 17,6 x 10-9 x 3003
= 43,76 J/mol K
Pr = 10/50,5 = 0,20
Tr = 300/283 = 1,06
De la carta de Edmister’s, Cp – Cp° @ 4 J/mol K
Así, Cp = 43,76 + 4 = 47,76 J/mol K
5.7. ENTALPÍA DE MEZCLAS
Para gases, los calores de mezcla son usualmente despreciables y las capacidades caloríficas y entalpías pueden tomarse como aditivas sin introducir errores significativos en los cálculos de diseño;
Cp(mezcla) = xa Cpa + xb Cpb + xc Cpc (5.20)
donde: xa , xb y xc son las fracciones molares de los componentes a, b y c.
Para mezclas de líquidos y para soluciones, el calor de mezcla (calor de disolución) puede ser significativo, y por lo tanto debe incluir al calcular las entalpías de la mezcla.
Para mezclas binarias, la entalpía específica de la mezcla a temperatura t está dada por:
Hmezcla = xa Ha,t + xb Hb,t + DHm,t (5.21)
donde: Ha,t , Hb,t son las entalpías específicas de los componentes a y b, y DHm,t es el calor de mezcla cuando se forma 1 mol de solución a la temperatura t.
Los calores de mezcla y calores de solución son determinados experimentalmente y están publicados para las soluciones más comunes.
Para soluciones orgánicas, el calor de mezcla, es usualmente pequeño comparado con otras cantidades de calor y puede despreciarse cuando se hacen los balances de calor para determinar los requerimientos de calentamiento o enfriamiento del proceso.
Los calores de solución de compuestos orgánicos e inorgánicos en agua pueden ser grandes, particularmente para los componentes fuertemente ácidos o alcalinos
5.7.1. Calores de solución integrales
Los calores de solución son dependientes de las concentraciones. El calor integral de solución a cualquier concentración dada es el total de calor liberado o absorbido durante la preparación de la solución a partir de solvente y soluto puros. El calor integral de solución a dilución infinita es denominado calor integral estándar de solución.
Tablas del calor integral de solución para un rango de concentración, y gráficas del calor integral de solución como una función de la concentración, son reportados en los “handbooks” para muchos de los diferentes materiales para los cuales el calor de solución es significante en cálculos de diseño de procesos.
El calor integral de solución se puede usar para calcular el calentamiento o enfriamiento requerido en la preparación de soluciones, como se ilustra en el Ejemplo 5.5.
Ejemplo 5.5
Una solución de NaOH en agua es preparada por la dilución de una solución concentrada en un tanque enchaquetado y con agitación. El poder de la solución concentrada es 50 por ciento p/p y 2 500 kg de solución al 5 por ciento p/p son requeridos por “batch”. Calcular el calor removido por el agua de enfriamiento si la solución debe ser descargada a una temperatura de 25 °C. La temperatura de la solución alimentada al tanque se puede tomar como 25 °C.
Solución
Calor integral de solución de NaOH – H2O, a 25 °C.
| Moles H2O/mol de NaOH | –DH°soluc kJ/mol NaOH |
| 2 | 22,9 |
| 4 | 34,4 |
| 5 | 37,7 |
| 10 | 42,5 |
| Infinito | 42,9 |
Conversión de porcentaje en peso a mol/mol:
50 por ciento p/p = 50/18 ¸ 50/40 = 2,22 mol H2O/mol de NaOH
5 por ciento p/p = 95/18 ¸ 5/40 = 42,2 mol H2O/mol de NaOH
De una gráfica de los calores integrales de solución “versus” la concentración.
– DH°soluc 2,22 mol/mol = 27,0 kJ/mol NaOH
42,2 mol/mol = 42,9 kJ/mol NaOH
Calor liberado en la dilución por mol de NaOH
42,9 – 27,0 = 15,9 kJ
Calor removido por “batch” = mol NaOH por “batch” x 15,9
![]()
Calor transferido al agua de enfriamiento, despreciando las pérdidas de calor,
= 49,7 MJ por “batch”
En el Ejemplo 5.5 las temperaturas de la solución final y la alimentación se han tomado iguales a la temperatura estándar para el calor de solución, 25 °C, para simplificar los cálculos. Los calores de solución son análogos a los calores de reacción, y ejemplos de balances de calor en procesos donde la temperatura es diferente de la temperatura estándar son dados en la discusión de calores de reacción, Sección 5.9.
5.8. DIAGRAMAS DE ENTALPÍA CONCENTRACIÓN
La variación de la entalpía para mezclas binarias son convenientemente representadas en diagramas. Un ejemplo común es el diagrama que muestra la entalpía de mezclas de amoniaco y agua en función de las concentraciones, con la presión y la temperatura como parámetros y publicada en la mayoría de textos de termodinámica. Esta cubre los cambios de fase de sólido a liquido y a vapor, y los valores dados de la entalpía incluyen los calores latentes para las fases de transición.
La entalpía es por kg de mezcla (amoniaco + agua)
Estado de referencia: entalpía del amoniaco a – 77 °C = cero
entalpía del agua a 0 °C = cero
Los diagramas de entalpía concentración facilitan los cálculos de los balances de energía involucrando cambios en la concentración y la fase.
Ejemplo 5.6.
Calcular la temperatura máxima cuando amoniaco liquido a 40 °C se disuelve en agua a 20 °C hasta formar una solución al 10 por ciento.
Solución
La máxima temperatura se alcanzará cuando no hay pérdidas de calor (proceso adiabático). Mientras no sea removido materia o calor el problema se puede resolver gráficamente en el diagrama concentración entalpía. La operación de mezcla se puede representar por un diagrama uniendo los puntos A representando amoniaco puro a 40 °C con el punto B representando agua pura a 20 °C. El valor de la entalpía de la mezcla cae sobre una línea vertical para la concentración requerida 0,1. La temperatura de la mezcla está dada por la intersección de esta línea vertical con la línea AB. Este método es una aplicación de la “regla de la palanca” para diagramas de fase. El método de Ponchon Savarit usado en el diseño de columnas de destilación, es un buen ejemplo de la aplicación de la regla de la palanca, y el uso de los diagramas entalpía concentración.

5.9. CALORES DE REACCIÓN
Si un proceso involucra reacción química, esta estará acompañada de una liberación o absorción de calor. La cantidad de calor depende de las condiciones bajo las cuales se lleva a cabo la reacción. El calor estándar de reacción es el calor liberado cuando la reacción es llevada a cabo bajo condiciones estándar: componentes puros, presión de 1 atm. (1,01325 bar), temperatura usualmente pero no necesariamente de 25 oC.
El estado de los reactantes y productos (gas, liquido o sólido) se debe especificar, si las condiciones de reacción son tales que ellas puedan existir en mas de un estado, por ejemplo:
H2(g) + ½ O2(g) ———-> H2O(g), DHro = – 241,6 kJ
H2(g) + ½ O2(g)————> H2O(l), DHro = – 285,6 kJ
La diferencia entre los dos calores de reacción es el calor latente del agua formado.
En cálculos de diseño de procesos, es usualmente más conveniente expresar el calor de reacción en términos de los moles de producto formado, para las condiciones bajo las cuales se lleva a cabo la reacción, kJ/mol de producto.
Los calores estándar de reacción se pueden convertir a otras temperaturas de reacción haciendo un balance de calor para un proceso hipotético, en el cual los reactantes son llevados a la temperatura estándar, la reacción llevada a cabo, y los productos son llevados luego a la temperatura de reacción requerida como se ilustra en la Fig. 5.2.
DHr,t = DHr° + DHprod – DHreaxt (5.22)

Fig. 5.2 DHr° a temperatura t
donde DHr,t = calor de reacción a temperatura t
DHreaxt = cambio de entalpía de los reactantes a la temperatura estándar
DHprod = cambio de entalpía de los productos a la temperatura estándar
Para reactores prácticos, donde los reactantes y productos pueden estar a temperaturas diferentes de la temperatura de reacción, es mejor llevar a cabo el balance de calor sobre la reacción actual usando la temperatura estándar de 25 °C como la temperatura de referencia; el calor de reacción estándar puede entonces usarse sin ninguna corrección.
Debe recalcarse que es innecesario hacer una corrección con respecto a la temperatura para el calor de reacción cuando se quiere realizar un balance de calor. Para un reactor práctico, el calor adicionado (o removido) Qp para mantener la temperatura de diseño del reactor estará dada por
Qp = Hproductos – Hreactantes – Qr (5.23)
Donde Hproductos es la entalpía total de la corriente de productos, incluyendo material no reaccionado y subproductos, evaluados a temperatura de 25 °C.
Hreactantes es la entalpía total de la corriente de alimentación, incluyendo exceso de reactantes e inertes, evaluada a temperatura de 25 °C.
Qr es el calor total generado por la reacción, evaluado a partir del calor estándar de reacción a 25 °C (298 K).
Qr = S – DHr x (mol de producto formado) (5.24)
Donde – DHr es el calor estándar de reacción por mol de un producto específico.
Nota: un signo negativo es necesario en la ecuación 5.24 así Qr es positivo cuando la reacción desprende calor, mientras que el cambio de entalpía estándar será negativa para reacciones exotérmicas. Qp será negativo cuando se requiere enfriamiento.
5.9.1. Efecto de la presión sobre el calor de reacción
La Ecuación 5.22 puede escribirse en una forma más general

Si el efecto de la presión es significante, el cambio en la entalpía de los reactantes y productos, desde las condiciones estándar se puede evaluar para incluir ambos efectos ( por ejemplo usando valores tabulados de la entalpía) y la corrección se realiza siguiendo el mismo camino que para la temperatura.
Ejemplo 5.7
Ilustra los cálculos manuales de un balance de calor en un reactor
Cloruro de vinilo (VC) es manufacturado mediante la pirolisis de 1,2 dicloroetano (DCE). La reacción es endotérmica. Los flujos para producir 5000 kg/h a 55 por ciento de conversión son mostrados en el diagrama. Ver ejemplo 4.13
El rector es un reactor tubular calentando gas combustible cuyo poder calorífico es de 33,5 MJ/m3.
Estimar la cantidad requerida de gas combustible.

Solución
![]()
La pequeña cantidad de impurezas, menos de 1 por ciento, que podrían estar presentes en la alimentación se han despreciado para el propósito de este ejemplo. También el rendimiento de VC ha sido tomado como 100 por ciento. La conversión se encuentra en el rango de 55 a 99 por ciento.
Datos de capacidad calorífica para la fase vapor.
CP° = a + bT + cT2 + dT3 kj/kmol K
| A | b x 102 | c x 105 | d x 109 | |
| VC | 5,94 | 20,16 | –15,34 | 47,65 |
| HCl | 30,28 | –0,761 | 1,325 | – 4,305 |
| DCE | 20,45 | 23,07 | –14,36 | 33,83 |
Para la fase liquida : DCE a 20 °C,
Cp = 116 kJ/kmol K, tomado como constante para el rango de temperatura entre 20 a 25 °C.
Calor latente de vaporización del DCE a 25 °C = 34,3 MJ/kmol
A 2 bar de presión, el cambio en el Cp con la presión es pequeño y puede despreciarse.
Tomando como temperatura base 25 °C (298 K), el estado estándar para DHr°
Entalpía de la alimentación = 145,5 x 116(293 – 298) = – 84,39 kJ/h = – 84,4 MJ/h
![]()
| Componente | ni mol/h | nia | nib x 103 | nic x 105 | nid x 109 |
| VC | 80 | 475,2 | 1612,8 | – 1227,2 | 3812,0 |
| HCL | 80 | 2422,4 | – 60,88 | 106,0 | – 344,4 |
| DCE | 65,5 | 1339,5 | 1511,0 | – 940,6 | 2215,9 |
| SniCp | 4237,1 | 3063,0 | – 2061,8 | 5683,5 |
![]()
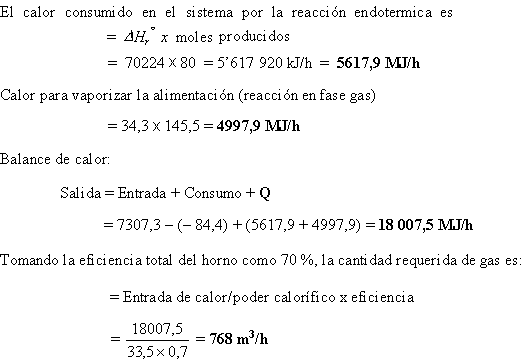
5.10. CALORES ESTÁNDAR DE FORMACIÓN
La entalpía estándar de formación DHfo de un compuesto se define como el cambio de entalpía cuando 1 mol de compuesto es formado desde sus elementos constituyentes en el estado estándar. La entalpía de formación de los elementos es tomada como cero. El calor estándar de reacción puede calcularse a partir de los calores de formación de los productos y reactantes; si estos están disponibles o pueden estimarse,
DHro = SDHfoproductos – SDHforeactantes (5.26)
5.11. CALORES DE COMBUSTIÓN
El calor de combustión de un compuesto DHco es el calor estándar de reacción para combustión completa del compuesto con oxígeno. Los calores de combustión son relativamente fáciles de determinarse experimentalmente. Los calores de otras reacciones pueden ser fácilmente calculados a partir de los calores de combustión de los reactantes y productos. La expresión general para el cálculo de calores de reacción a partir de calores de combustión es:
DHro = SDHcoreactantes – SDHcoproductos (5.27)
Nota: los términos producto y reactante están en dirección contraria a la que está dada en la expresión para el cálculo a partir de los calores de formación.
Los calores de combustión son grandes comparados con los calores de reacción.
5.12. COMPRESIÓN Y EXPANSIÓN DE GASES
El término trabajo en un balance de energía es improbable sea cero cuando un gas es expandido o comprimido como parte del proceso. Al computar el trabajo de presión con el término
![]()
es necesaria una relación entre la presión y el volumen durante la expansión.
Si la compresión o expansión es isotérmica (a temperatura constante), entonces por unidad de masa de gas con comportamiento ideal:
P v = constante (5.28)
y el trabajo es:
![]()
donde: P1 = presión inicial
P2 = presión final
v1 = volumen inicial
En compresores o expansores industriales la trayectoria de la compresión o expansión será “politrópica”, dada aproximadamente por la expresión:
P vn = constante (5.30)
El trabajo producido (o requerido) está dado por la expresión general:

donde: Z = factor de compresibilidad (1 para comportamiento ideal)
R = constante universal de los gases 8,314 J oK-1 mol-1
T1 = temperatura de entrada oK
M = masa molecular (peso) del gas.
El valor de n dependerá del diseño y operación de la máquina
La energía requerida para comprimir un gas, o la energía obtenida de la expansión, se estima mediante el cálculo del trabajo ideal y aplicando una adecuada eficiencia
5.14 BALANCES DE ENERGÍA AL ESTADO NO ESTACIONARIO
Todos los ejemplos de balance de energía considerados previamente han sido para procesos al estado estacionario; en los cuales la cantidad de generación o consumo de energía no varía con el tiempo y el término de acumulación en la ecuación general de balance de energía es tomada como cero.
Si se considera un proceso “batch”, o si la cantidad de energía generada o removida varía con el tiempo, será necesario establecer un balance diferencial de energía, similar al balance diferencial de materiales considerado en el Capítulo 4. Para procesos “batch” los requerimientos totales de energía pueden usualmente estimarse tomando 1 “batch” como la base de cálculo, pero la máxima velocidad de generación de calor también deberá estimarse para dimensionar cualquier equipo necesario de transferencia de calor.
Ejemplo 5.9
Balance diferencial de energía. En el proceso de separación “batch” de una solución acuosa, es preciso calentar el agua hasta 80 oC en un recipiente agitado y enchaquetado; 1 000 gal Imp. (4 545 kg) son calentados desde 15 oC. Si el área de la chaqueta es 300 pies2 (27 m2) y el coeficiente total de transferencia de calor se puede tomar como 50 Btu/h.pie2.oF (285 W/m2.oK), estimar el tiempo de calentamiento. El vapor suministrado es a 25 psig (2,7 bar)
Solución
La velocidad de transferencia de calor desde la chaqueta al agua está dada por la expresión:
![]()
donde: dQ = es la cantidad de calor transferido en el intervalo de tiempo dt
U = coeficiente total de transferencia de calor
Ts = temperatura de saturación del vapor
T = temperatura del agua
El incremento de temperatura del agua dT, se relaciona con el calor transferido dQ mediante la ecuación de balance de energía
dQ = w Cp dT (b)
donde: w Cp es capacidad calorífica del sistema.
Igualando las ecuaciones (a) y (b).

Tiempo de calentamiento por “batch”

En este ejemplo se ha despreciado la capacidad calorífica del recipiente y las pérdidas de calor para simplificar los cálculos. Ellos deberían incrementar el tiempo de calentamiento en 10 a 20 %.